Analysis immune repertoire kmer statistics: sequence profiles, etc.
Source:R/v0_kmers.R
split_to_kmers.Rd![[Deprecated]](figures/lifecycle-deprecated.svg)
Usage
split_to_kmers(.data, .k)
kmer_profile(.data, .method = c("freq", "prob", "wei", "self"), .remove.stop = TRUE)Arguments
- .data
Character vector or the output from
getKmers.- .k
Integer. Size of k-mers.
- .method
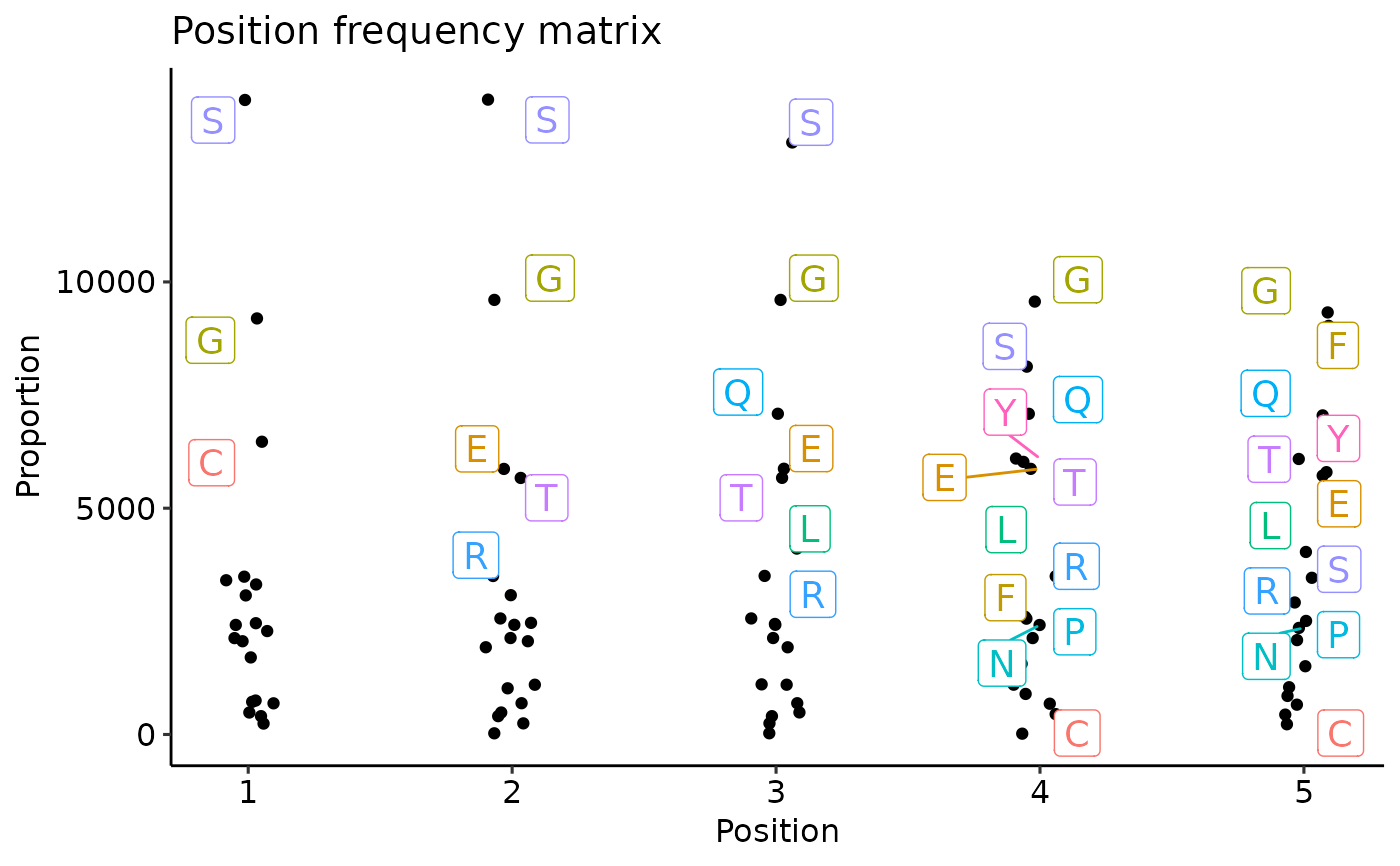
Character vector of length one. If "freq" then returns a position frequency matrix (PFM) - a matrix with occurences of each amino acid in each position.
If "prob" then returns a position probability matrix (PPM) - a matrix with probabilities of occurences of each amino acid in each position. This is a traditional representation of sequence motifs.
If "wei" then returns a position weight matrix (PWM) - a matrix with log likelihoods of PPM elements.
If "self" then returns a matrix with self-information of elements in PWM.
For more information see https://en.wikipedia.org/wiki/Position_weight_matrix.
- .remove.stop
Logical. If TRUE (by default) remove stop codons.
Value
split_to_kmers - Data frame with two columns (k-mers and their counts).
kmer_profile - a matrix with per-position amino acid statistics.
Examples
data(immdata)
kmers <- getKmers(immdata$data[[1]], 5)
kmer_profile(kmers) %>% vis()
#> Warning: Warning: removed 5 non-amino acid symbol(s): A
#> Please make sure your data doesn't have them in the future.
#> Warning: Removed 5 rows containing missing values or values outside the scale range
#> (`geom_point()`).
#> Warning: Removed 5 rows containing missing values or values outside the scale range
#> (`geom_label_repel()`).
#> Warning: ggrepel: 20 unlabeled data points (too many overlaps). Consider increasing max.overlaps