![[Deprecated]](figures/lifecycle-deprecated.svg)
An utility function to visualise the output from repClonality().
Arguments
- .data
Output from
repClonality().- .by
Pass NA if you want to plot samples without grouping.
You can pass a character vector with one or several column names from ".meta" to group your data before plotting. In this case you should provide ".meta".
You can pass a character vector that exactly matches the number of samples in your data, each value should correspond to a sample's property. It will be used to group data based on the values provided. Note that in this case you should pass NA to ".meta".
- .meta
A metadata object. An R dataframe with sample names and their properties, such as age, serostatus or hla.
- .errorbars
A numeric vector of length two with quantiles for error bars on sectors. Disabled if ".errorbars.off" is TRUE.
- .errorbars.off
If TRUE then plot CI bars for distances between each group. Disabled if no group passed to the ".by" argument.
- .points
A logical value defining whether points will be visualised or not.
- .test
A logical vector whether statistical tests should be applied. See "Details" for more information.
- .signif.label.size
An integer value defining the size of text for p-value.
- ...
Not used here.
Details
If data is grouped, then statistical tests for comparing means of groups will be performed, unless .test = FALSE is supplied.
In case there are only two groups, the Wilcoxon rank sum test (https://en.wikipedia.org/wiki/Wilcoxon_signed-rank_test) is performed
(R function wilcox.test() with an argument exact = FALSE) for testing if there is a difference in mean rank values between two groups.
In case there more than two groups, the Kruskal-Wallis test (https://en.wikipedia.org/wiki/Kruskal%E2%80%93Wallis_one-way_analysis_of_variance) is performed (R function kruskal.test()), that is equivalent to ANOVA for ranks and it tests whether samples from different groups originated from the same distribution.
A significant Kruskal-Wallis test indicates that at least one sample stochastically dominates one other sample.
Adjusted for multiple comparisons P-values are plotted on the top of groups.
P-value adjusting is done using the Holm method (https://en.wikipedia.org/wiki/Holm%E2%80%93Bonferroni_method) (also known as Holm-Bonferroni correction).
You can execute the command ?p.adjust in the R console to see more.
Examples
# \dontrun{
data(immdata)
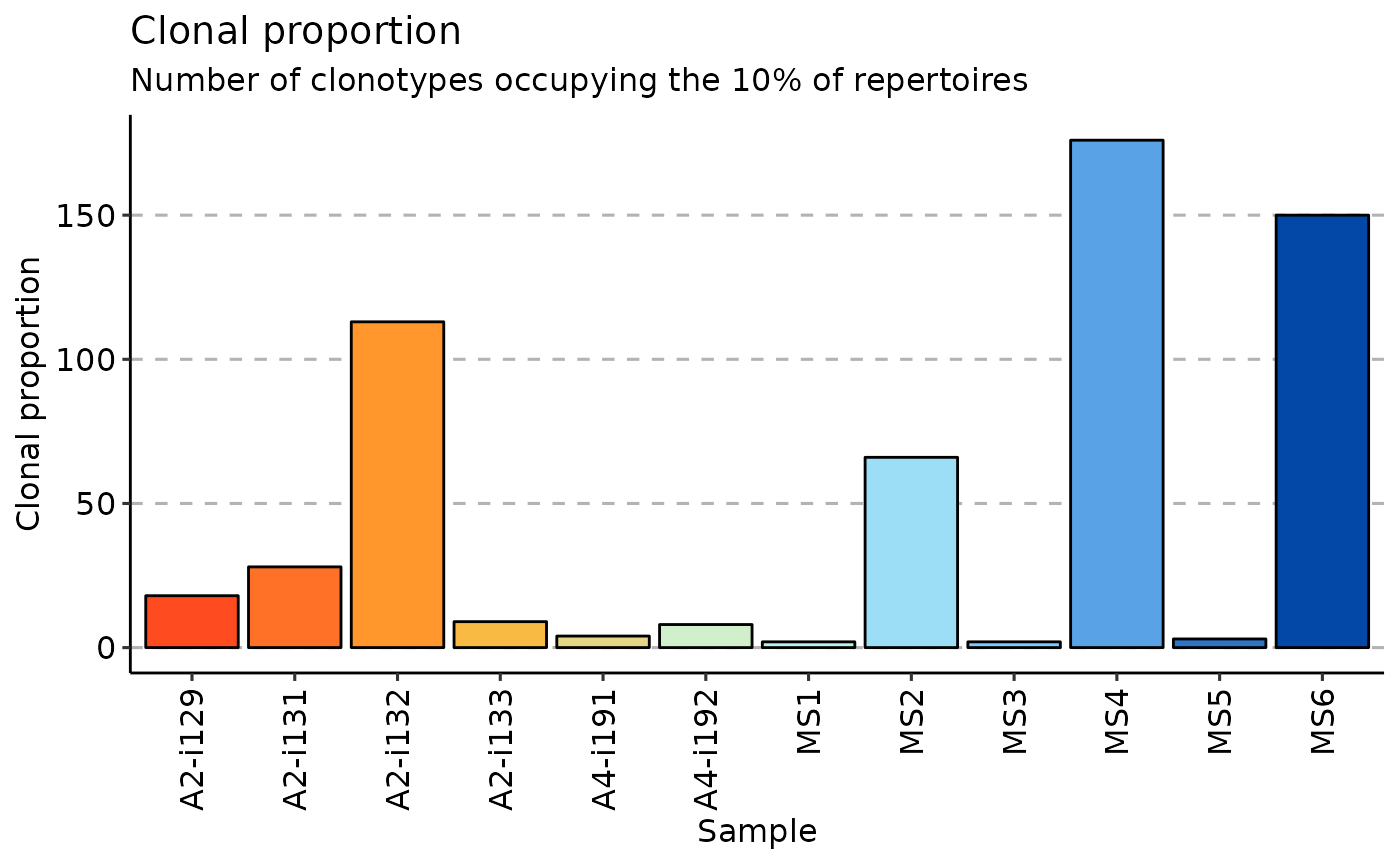
clp <- repClonality(immdata$data, "clonal.prop")
vis(clp)
 hom <- repClonality(immdata$data, "homeo")
# Remove p values and points from the plot
vis(hom, .by = "Status", .meta = immdata$meta, .test = FALSE, .points = FALSE)
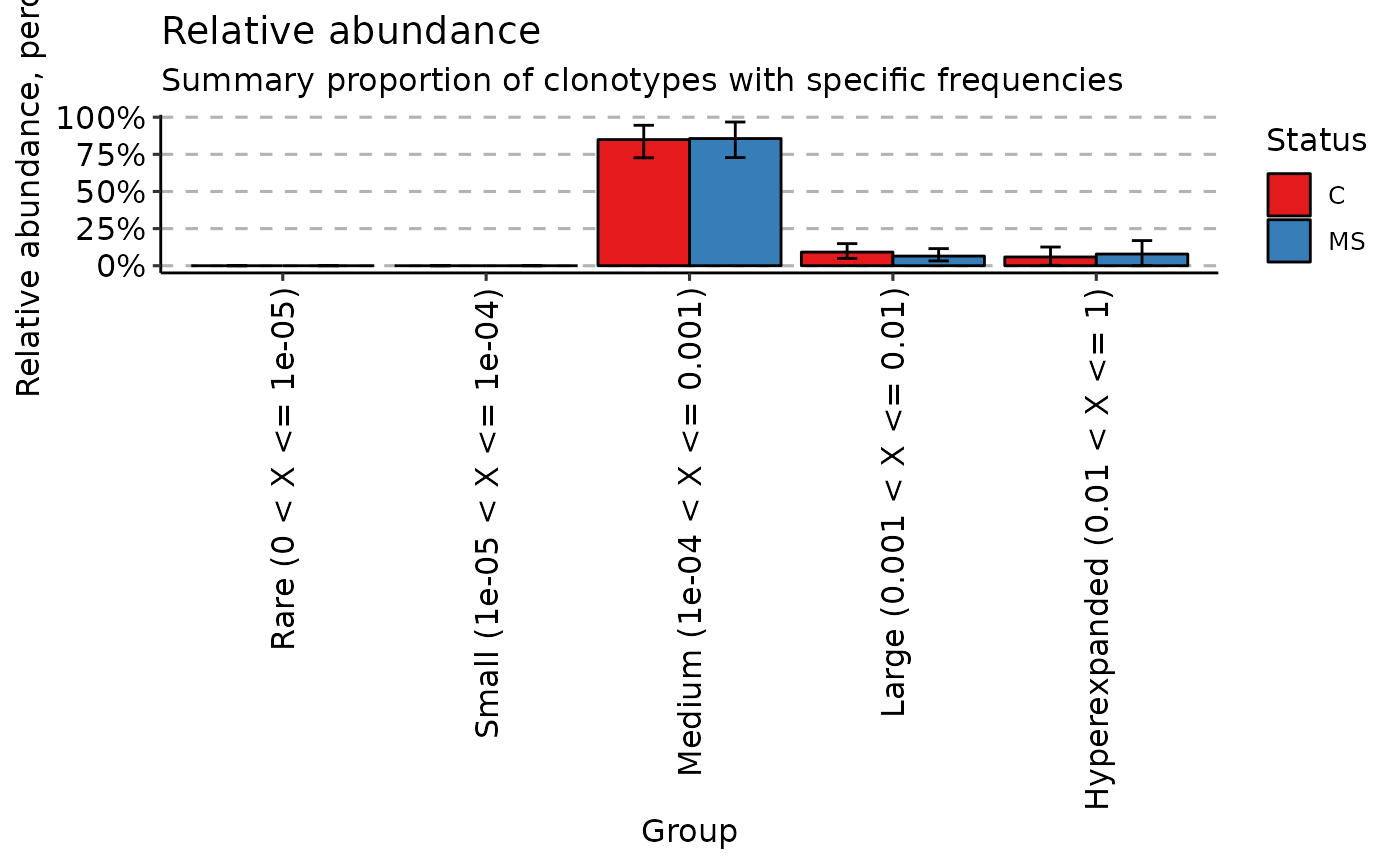
hom <- repClonality(immdata$data, "homeo")
# Remove p values and points from the plot
vis(hom, .by = "Status", .meta = immdata$meta, .test = FALSE, .points = FALSE)
 # }
# }