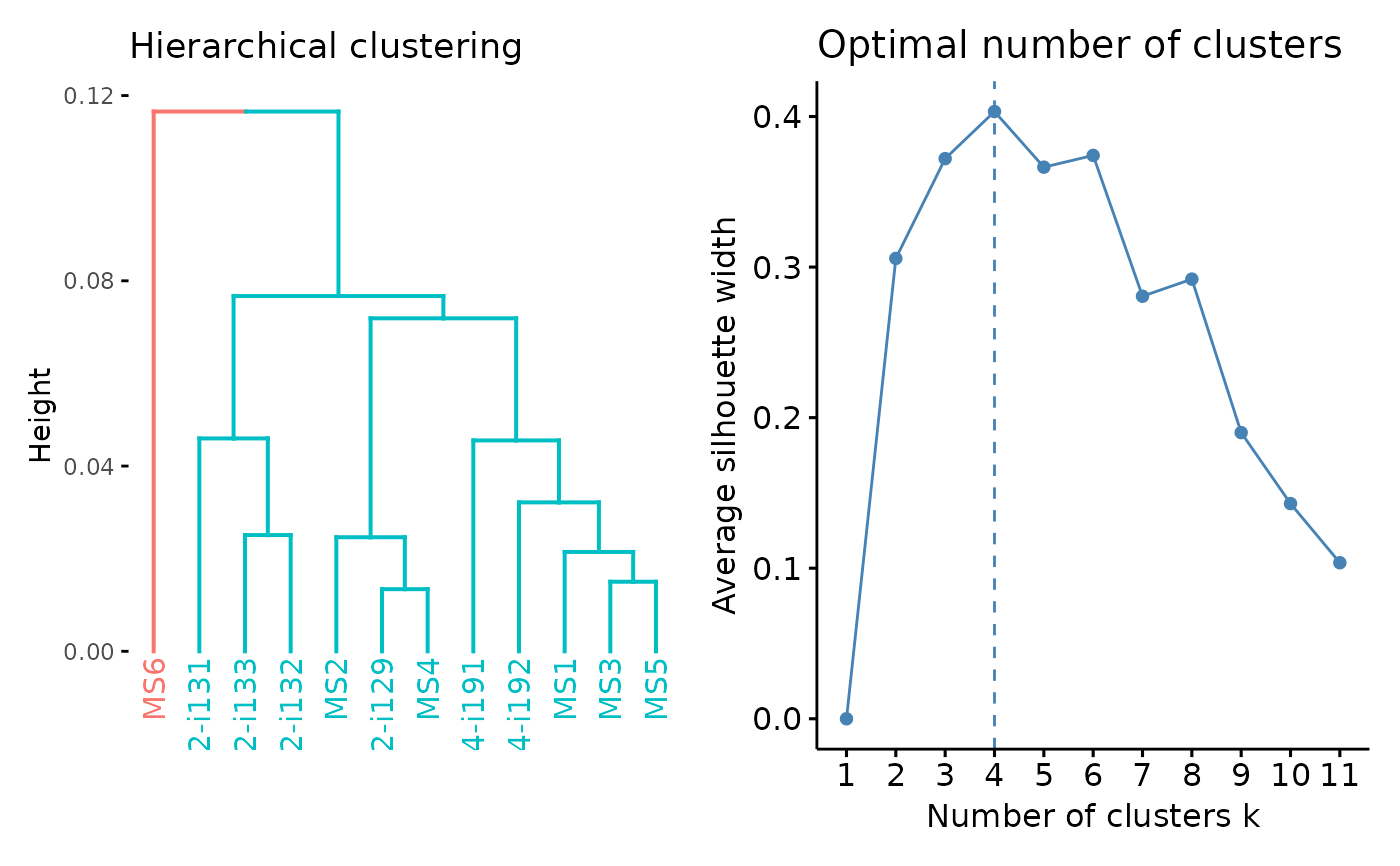
Post-analysis of V-gene and J-gene statistics: PCA, clustering, etc.
Source:R/v0_gene_usage_analysis.R
geneUsageAnalysis.Rd![[Deprecated]](figures/lifecycle-deprecated.svg)
The geneUsageAnalysis() function deploys several
data analysis methods, including PCA, multidimensional scaling,
Jensen-Shannon divergence, k-means, hierarchical clustering, DBscan, and different
correlation coefficients.
Arguments
- .data
The
geneUsageAnalysis()function runs on the output fromgeneUsage().- .method
A string that defines the type of analysis to perform. Can be "pca", "mds", "js", "kmeans", "hclust", "dbscan" or "cor" if you want to calculate correlation coefficient. In the latter case you have to provide
.corargument.- .base
A numerical value that defines the logarithm base for Jensen-Shannon divergence.
- .norm.entropy
A logical value. Set TRUE to normalise your data if you haven't done it already.
- .cor
A string that defines the correlation coefficient for analysis. Can be "pearson", "kendall" or "spearman".
- .do.norm
A logical value. If TRUE it forces Laplace smoothing, if NA it checks if smoothing is necessary, if FALSE does nothing.
- .laplace
The numeric value, which is used as a pseudocount for Laplace smoothing.
- .verbose
A logical value.
- .k
The number of clusters to create, passed as
kto hcut or ascentersto kmeans.- .eps
A numerical value, DBscan epsylon parameter, see
immunr_dbscan().- .perp
A numerical value, t-SNE perplexity, see
immunr_tsne().- .theta
A numerical value, t-SNE theta parameter, see
immunr_tsne().
Value
Depends on the last element in the .method string. See immunr_tsne for more info.
Examples
data(immdata)
gu <- geneUsage(immdata$data, .norm = TRUE)
geneUsageAnalysis(gu, "js+hclust", .verbose = FALSE) %>% vis()
#> Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
#> ℹ Please use tidy evaluation idioms with `aes()`.
#> ℹ See also `vignette("ggplot2-in-packages")` for more information.
#> ℹ The deprecated feature was likely used in the factoextra package.
#> Please report the issue at <https://github.com/kassambara/factoextra/issues>.
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the factoextra package.
#> Please report the issue at <https://github.com/kassambara/factoextra/issues>.
#> Warning: The `<scale>` argument of `guides()` cannot be `FALSE`. Use "none" instead as
#> of ggplot2 3.3.4.
#> ℹ The deprecated feature was likely used in the factoextra package.
#> Please report the issue at <https://github.com/kassambara/factoextra/issues>.